Quantum chemists have often hoped that it may be possible to calculate the ground state energy of an atom or a molecule without the need for traditional wavefunction-based methods. One method, first proposed in the 1950s, requires variational calculations on so-called second-order reduced density matrices. However it wasn’t until the late 1990s that scientists realized that this approach is equivalent to a well-known problem called a semidefinite program, and can be solved using optimization software on parallel computers.
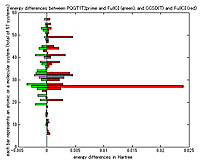
Since 2001, Mituhiro Fukuda of Tokyo Tech’s Department of Mathematical and Computing Sciences, Maho Nakata of RIKEN, Bastiaan J. Braams of AIEA, Jerome K. Percus of New York University and other co-workers have been imposing different ‘N-representability’ conditions on the density matrices to obtain tighter approximations for the ground state energies. In particular, computations imposing certain conditions called P, Q, G, T1, and T2’ seem to be comparable to using the CCSD(T) method, which is generally considered to be the gold-standard in quantum chemistry. The largest system they solved was the H2O molecule in the so-called 1A1 state; this gave an energy just 0.4 miliHartree lower than the full configuration interaction calculation, whereas CCSD(T) gave a difference of 0.55 miliHartree.
Unfortunately, this method is unlikely to compete with traditional methods in terms of computational time and size. However, computations on so-called one-dimensional Hubbard models using an ultra-precise semidefinite programming solver have shown that the method of Fukuda and co-workers is a promising approach to models where traditional methods produce computational failures.
Reference
- Authors: Maho Nakata, Bastiaan J. Braams, Katsuki Fujisawa, Mituhiro Fukuda, Jerome K. Percus, Makoto Yamashita, and Zhengji Zhao.
- Title of original paper: Variational calculation of second-order reduced density matrices by strong N-representability conditions and an accurate semidefinite programming solver.
- Journal, volume, pages and year: Journal of Chemical Physics 128, 164113 (2008).
- Digital Object Identifier (DOI): 10.1063/1.2911696
- Affiliations: RIKEN, AIEA, Chuo University, Department of Mathematical and Computing Sciences, New York University, LBNL.
- Department website: http://www.is.titech.ac.jp/index-e.html